

Actualités
Partenaires

Étude ab initio de la structure électronique de la réaction N (2D) + CH4, d’ intérêt pour la physico-chimie de l’atmosphère de Titan
Chanda-Malis OUK
Laboratoire UTINAM, équipe DREAM
16 Route de Gray, 25000 Besançon
Vue d’artiste de l’entrée de la sonde Huygens le 14/01/2005
dans l’atmosphère de Titan, le plus gros satellite de Saturne :

De par son abondance dans les atmosphères terrestre et planétaires, l'atome d'azote N joue un rôle majeur dans les réactions chimiques atmosphériques. En effet, les composés moléculaires les plus abondants détectés dans l'atmosphère de Titan (Satellite de Saturne) sont N2 (94%) et CH4 (2 à 5%). On observe ainsi la présence de nitriles qui laissent supposer l'existence de mécanismes réactionnels impliquant l'azote atomique ou ionique formé par dissociation ou ionisation de N2.
Depuis plusieurs années, les réactions impliquant l'atome d'azote dans son premier état excité N(2D) ont fait l'objet d'un certain nombre d'études [1-4] qui se révèlent erronées de nos jours.
Une étude expérimentale récente de la réaction N (2D) + CH4 par une technique de faisceaux moléculaires croisés (Crossed Molecular Beam)a permis de caractériser les produits issus des nombreuses voies de sortie thermodynamiquement possibles (cf Figure 1)
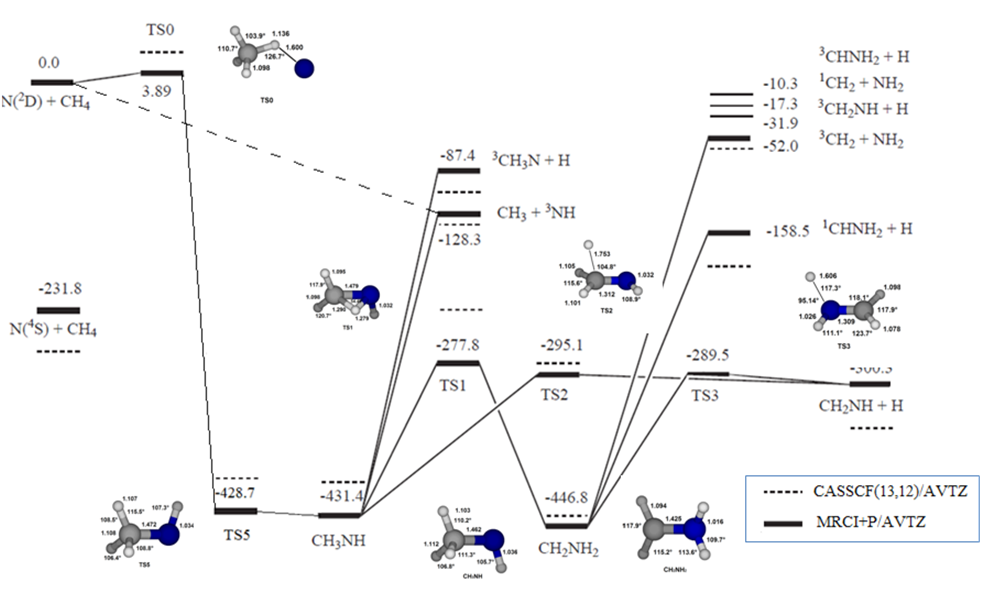
Figure 1. Diagramme énergétique de la surface d'énergie potentielle de N(2D) + CH4. Énergies en kJ/mol.
L’étude ab initio de cette réaction se montre particulièrement couteuse en temps de calcul du fait de la grande dimensionnalité du système: 6 atomes et 12 degrés de liberté. Calculer seulement 4 arrangements moléculaires par degré de liberté nécessiterait de calculer 412 = 16.7 millions de points ce qui n’est pas réalisable de nos jours en un temps raisonnable pour des calculs ab initio. D’autre part, nous avons montré récemment la nécessité d’inclure la corrélation électronique à un haut niveau de calcul afin d’obtenir des taux de réaction en accord avec les valeurs expérimentales.
Le Grand Challenge 2012 nous a ainsi permis de calculer plus de 4000 arrangements géométriques de notre système au meilleur niveau de corrélation possible (MRCI), sachant qu’un arrangement requière en moyenne 24H de temps cpu. Ce nombre d’arrangements reste encore insuffisant pour bien décrire toute la surface d’énergie potentielle globale mais constitue déjà une bonne base de travail que nous devons encore améliorée par la suite.
[1] Y. Kurosaki, T. Takayanagi, K. Sato, S.Tsunashima, J. Phys. Chem A 1998, 102, 254-259
[2] T. Takayanagi, Y. Kurosaki, K. Sato, K. Misawa, Y. Kobayashi, S. Tsunashima, J. Phys. Chem. A 1999, 103, 250-255
[3] T. Takayanagi, Y. Kurosaki, Journal of Molecular Structure (Theochem)1999, 492, 151-158
[4] T. Takayanagi, Y. Kurosaki, K. Yokoyama, International Journal of Quantum chemistry 2000, 79, 190-197
[5] N. Balucani, D. Skouteris, M. Rosi, J. Phys. Chem. A 2009, 113, 11138-11152.

