

Actualités
Partenaires

Traitement par DFT de systèmes moléculaires de très grandes tailles sans approximation
Eric Duverger
Institut FEMTO-ST, Département Micro Nano Sciences & Systèmes
Un des aspects fascinant actuellement en chimie supramoléculaire est la possibilité par ingénierie d'architectures de développer des molécules en fonction de leurs applications. On peut ainsi concevoir des molécules dont le mouvement moléculaire permettra d’exécuter des fonctions préprogrammées dans les appareils ou les matériaux. Pour déclencher de tels mouvements à l'échelle moléculaires, on cherche couramment à utiliser une réaction cis-trans stimulée par la lumière ou la chaleur. Dans le cadre de notre programme de recherche visant à la préparation d'architectures multi-composants avec l’Université Catholique de Louvain, nous étudions une molécule arachnoïde pouvant montrer un mouvement moléculaire de ses jambes azobenzenyl lors de stimulations lumineuses.
Des premiers résultats expérimentaux intéressants ont été obtenus en déposant une molécule isolée en interaction faible sur un substrat métallique ou de graphène. Toutefois, afin d’obtenir une compréhension complète du système (parties de la molécule impliquées dans le mouvement, positions etc..), une étude théorique se relève indispensable. Dans le cadre du Grand Challenge 2012, nous avons réalisé à l’aide d’un code de calcul parallèle full DFT (VASP) des simulations théoriques du système. Cette approche théorique était un véritable challenge compte tenu de la taille latérale des systèmes et du nombre d’atome considéré (~8000 atomes, taille 7x7 nm2). Les résultats obtenus sont conformes à nos espérances :
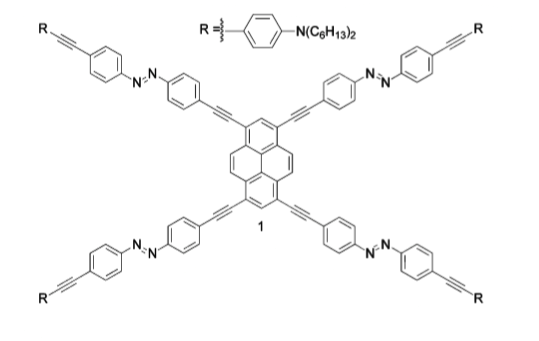
Figure 1: Molécule arachnoïde étudiée
Cette étude nous a permis de prouver la possibilité en HPC de traiter par DFT et en tenant compte de la relaxation, des systèmes de très grandes tailles sans approximation. Pour réaliser l’étude d’un tel système nous avons utilisé 120 processeurs avec 5 Go de mémoire vive par processeur. Après l’optimisation du système en relaxation complète avec son substrat de graphène, nous avons poursuivi cette première étude en calculant l’image STM correspondante. Le calcul a été réalisé en tenant compte d’une pointe virtuelle en tungstène. Les résultats théoriques confortent les résultats expérimentaux. Nous souhaitons poursuivre cette étude dans le futur pour réaliser une étude complète des résultats expérimentaux en fonction des différentes conformations observées sur la surface. L’ensemble des données obtenues sera ensuite intégré dans une communication en vue d’une publication internationale.

Figure 2 : Image STM de la Molécule arachnoïde

